我已经在计算生物领域工作了几个月,并且学到了很多东西。还有很多我不知道,但我目前正处于一个阶段,我将一些部分拼凑在一起,同时仍然记得不理解它们的感觉,这通常是尝试写介绍性内容的好时机.尝试解释事情也是确保我自己理解它们的好方法。所以,这里是我一直在学习的东西的转储:
生物世界主要用核酸存储信息。这些是一系列核苷酸,通常称为碱基: A 、 C 、 G和T。例如,一条核苷酸链可能看起来像:
GGCTGAGACAGTGCCCAGATCGTTACACCATTCGT
两种主要的核酸是DNA和RNA 。它们在一些方面有所不同,但从计算的角度来看,它们非常相似。物理 RNA 将使用U而不是 T,尽管在测序数据中您经常会看到它带有 T。
每个碱基都有一个互补物:A 与 T 键合,G 与 C 键合。包含两条键合链的核酸称为双链。一条链中的每个碱基都将与其互补链结合:
GGCTGAGACAGTGCCCAGATCGTTACACCATTCGT |||||||||||||||||||||||||||||||||| CCGACTCTGTCACGGGTCTAGCAATGTGGTAAGCA
这是著名的双螺旋结构,比单链核酸更稳定。
从物理核酸到计算机上的序列是测序,反之则是合成。我只谈前者;我对后者了解不多。
当今最常见的测序方法是下一代测序,在主要供应商之后通常称为Illumina测序。底座被染色,机器读取它们的颜色。测序的输出是大量的短读。每次读取是 50-300 个碱基的序列,通常约为 150 个。在设置测序运行时,您可以选择要读取的碱基数,不同的应用程序将在不同的长度下最有意义。当您沿着链读得更远时,准确性会下降。请注意,我们所说的长度远小于完整核酸的长度,通常至少有数千个碱基。没有得到全貌是这种排序的一个很大的缺点。
让我们得到一些真实的数据来玩。当人们发表依赖于测序的论文时,他们通常会将原始数据上传到 NIH 的国家生物技术信息中心(NCBI)。这是我最近一直在看的一篇论文,它对废水进行了测序:南加州废水的 RNA 病毒学和 SARS-CoV-2 Single-Nucleotide Variants 的检测。如果您查看“数据可用性”部分,您会看到:
原始测序数据已以登录号PRJNA729801保存在 NCBI 序列读取存档中,代表性代码可在https://github.com/jasonarothman/wastewater_viromics_sarscov2找到。
GitHub 链接有助于获取元数据(每个样本代表什么?)并了解他们如何处理元数据(他们使用了哪些工具以及如何处理?),但现在我们正在寻找测序数据。登录号是“PRJNA729801”,虽然我们可以从 NCBI 中点击并下载,但欧洲镜像(European Nucleotide Archive)上的用户界面要好得多。我们转到他们的登录页面并输入登录号:
这会将我们带到描述数据的页面:
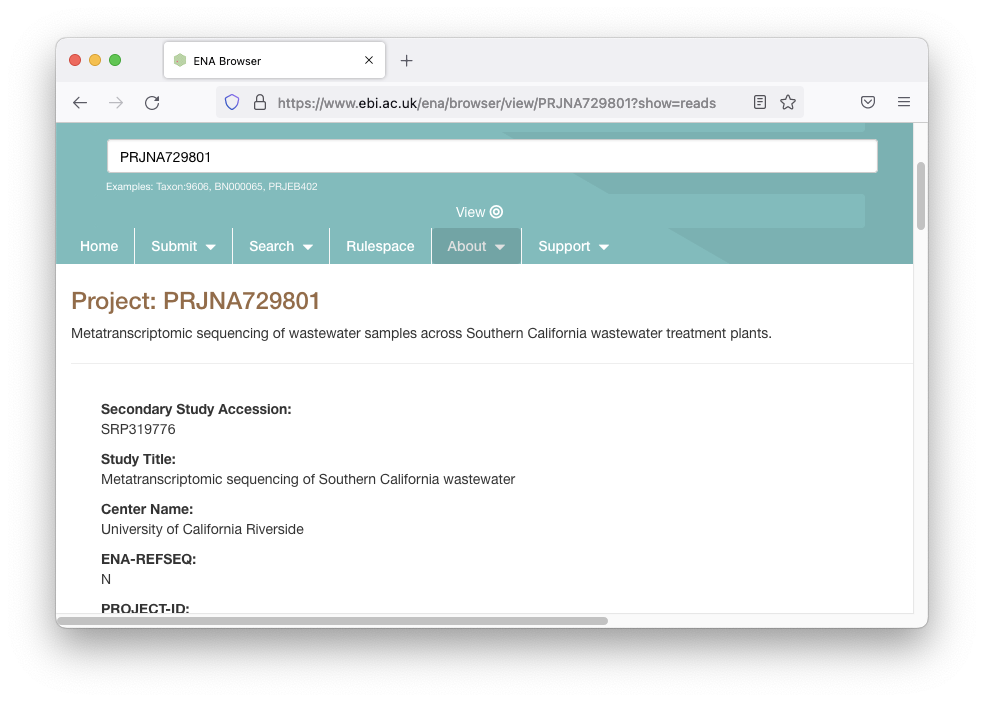
我们想要彻底检查标题,以确保我们最终没有得到错误的数据集,并且“南加州废水的元转录组测序”听起来是正确的。
向下滚动有链接:
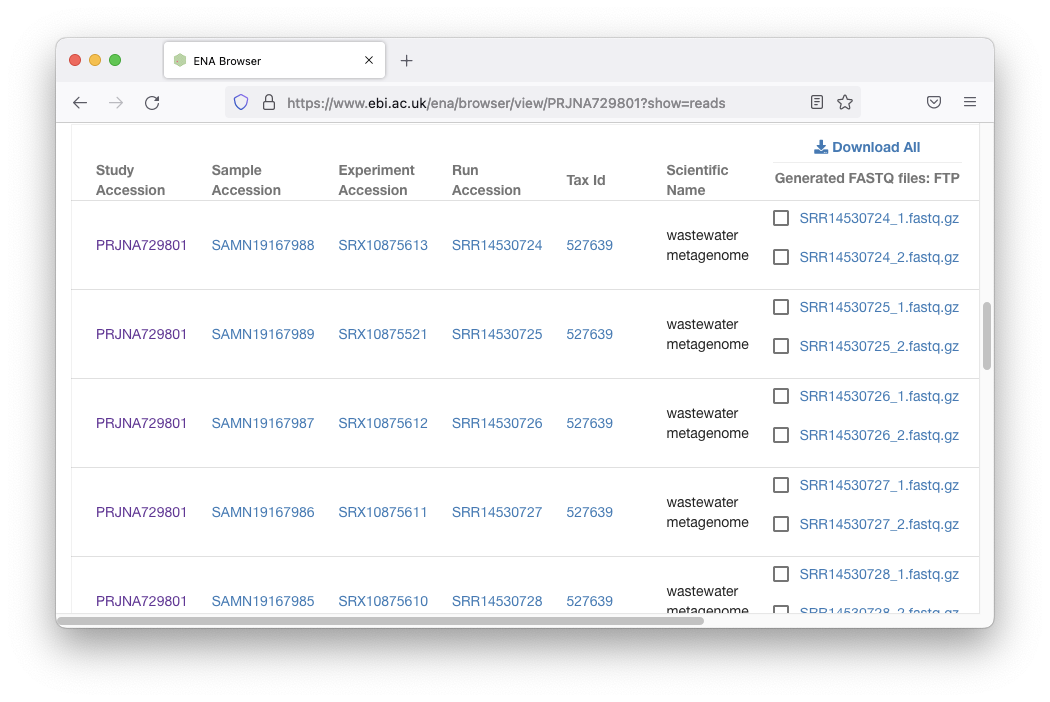
我们可以下载所有这些数据,但压缩后的大小约为 80GB。现在,让我们下载一个 ~150MB 的fastq.gz文件: SRR14530724_1.fastq.gz 。
这些文件通常都非常大且非常重复,因此它们是压缩的自然候选者。最常见的选项是 gzip,这就是他们在这里使用的。让我们从解压缩它开始:
$ gunzip SRR14530724_1.fastq.gz
现在我们可以看一下:
$头-n 4 SRR14530724_1.fastq @SRR14530724.1 1/1 GTTGTTATCCTGCGCGGCGACGCCAGCCTTGCTCAATTGCTCGAGCAGGGC CTGTCTCTTATACACATCTCCGAGCCCACGAGACAGGTCAGATAATCTCGT ATGCCGTCTTCTGCTTGAAAAGGGGGGGGGGGGGGGGGGGGGGGGGGGG + FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF F:F:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF,FFF FFF:FF,FFF::FF::F::::,FFFFFFFFFFFFFFFFFFFFFFFFFF
这种文件格式称为FASTQ ,现在我们正在查看文件的单次读取。以“@”开头的行给出了这个序列的 id,然后是序列本身。以“+”开头的行表示序列的结束,然后是质量分数,我们稍后会谈到。
我可以看到这个文件中有 270 万次读取:
$ grep -c ^@ SRR14530724_1.fastq 2737872
你会注意到这个读取以一长串“G”结尾,这在数据中实际上很常见:
$ grep -c 'GGGGGGGG$' SRR14530724_1.fastq 840839 # 31% 的读取
这称为Poly-G ,来自该测序仪使用的特定化学物质。它通过将 A 和 T 染成一种颜色(绿色)和 A 和 C 染成不同的颜色(红色)来识别碱基。这意味着 A 为黄色(绿色+红色灯),T 为绿色,C 为红色,G 为黑色(无灯)。测序仪无法区分“没有碱基”和“有碱基但它们都是G并且没有拾取任何染料”。当一个序列比预期的短时,它会得到一个 Poly-G 尾巴。作为一般质量控制步骤的一部分,您通常会在测序后修剪这些尾巴;我们现在只做sed 's/GGGGGGGG*$//' 。
定序器有一些关于它的自信程度的信息:有时它会看到一种清晰的颜色,有时会混合一些另一种颜色。这就是质量得分编码的内容。它与序列的长度相同,它们对应一个字符。
通过获取字符的 ASCII 值并减去第一个非空白可打印字符 (‘!’) 的 ASCII 值 33,将字母转换为数值。要将其转换为乘以 -0.1 的错误概率,请取 10 的幂。例如,’F’ 是 ASCII 70,而10^(-.1*(70-33))是 0.02% 的错误几率,或 99.98% 的准确几率。十个 ASCII 位置的增加表明精度提高了 10 倍。
此音序器仅使用几个不同的质量值:
| 字符 | 数值 | 准确性 |
|---|---|---|
| F | 37 | 99.98% |
| : | 25 | 99.7% |
| , | 11 | 92% |
| # | 2 | 37% |
看着这一次测序运行,我看到:
$ 猫 SRR14530724_1.fastq | grep FF # 仅质量行 | sed 's/\(.\)/\1\n/g' # 每行一个字符 | grep # 忽略空格 | head -n 10000000 # 对数据进行采样 |排序 | uniq -c # 计数类型 257 # # ~0% 411192 , # 4% 506844 : # 5% 9081707 F # 91%
让我们只提取序列的一小部分及其相应的质量分数:
CTGTCTCT F:F:FFFF
这就是说它对前两个“T”有 99.7% 的信心,对其他电话有 99.98% 的信心。
如果您不关心质量分数,您通常会使用“FASTA”文件格式:
>SRR14530724.1 1/1 GTTGTTATCCTGCGCGGCGACGCCAGCCTTGCTCAATTGCTCGAGCAGGGC CTGTCTCTTATACACATCTCCGAGCCCACGAGACAGGTCAGATAATCTCGT ATGCCGTCTTCTGCTTGAAAAGGGGGGGGGGGGGGGGGGGGGGGGGGGG >SRR14530724.2 2/1 CATTTTCGACGGCGTCGATGTACAAAGGTTATACCATAGTAAGTCCGAAGC TACAGGCTTATGACACCGCAGAGTCAATGTATTCCGGTGACAATGTACTGA TGTACAGTGGGACTGACACTGTCTCTTATACACATCTCCGAGCCCACGA
“>” 标记读取的开始,之后的所有内容都是该序列的 id。例如,该文件中的第一个读取是“SRR14530903.1 1/2”。直到下一个“>”之前的所有行都是该读取的序列。
您会注意到一些文件以_1.fastq.gz ,一些以_2.fastq.gz ,并且每一对都具有相同的读取次数:
$ grep -c ^@ SRR14530724_*.fastq SRR14530724_1.fastq:2737872 SRR14530724_2.fastq:2737872
这是因为这个特定的数据集使用了双端测序。这个想法是您同时对每个片段的开头部分(正向读取)和末尾部分(反向读取)进行排序,分别将它们放在_1和_2文件中。它们之间有一个未测序的间隙,您可以根据您在测序前如何切碎片段来了解它的长度。这对于弄清楚这些片段如何组装成完整的基因序列很有用。
到目前为止,所有这些都描述了短的 Illumina 式读取,但还有另一种越来越流行的测序,称为纳米孔测序。核酸通过芯片上的一个小孔送入,不同的碱基序列在通过时会产生不同的电信号。将这些信号转换为 ACTG 字符串并生成 FASTQ 文件是碱基调用,您通常使用在 GPU 上运行的神经网络。这产生比 Illumina 测序长得多的读数,因为您可以通过任意长度的核酸进料。数万个碱基的读取是常见的,并且数百万个碱基的读取是可能的。长读对很多事情都有帮助,尤其是当您使用从皮肤、唾液、废水等自然环境中提取的核酸混合物时(宏基因组学、元转录组学)。在这一点上,Nanopore 的成本与 Illumina 大致相当,但仍然有更高的错误率。
让我们在这里停下来,因为这已经足够长了。我可能会在某个时候写其他东西:组装、qPCR、扩增子测序、反向互补、工具、k-mers。很高兴回答问题!